Clinical Focus ›› 2021, Vol. 36 ›› Issue (5): 436-441.doi: 10.3969/j.issn.1004-583X.2021.05.010
Previous Articles Next Articles
Clinical characteristics of hereditary renal tubular disease in hypokalemia
Liu Fan1a, Sun Yan1a,2a( ), Xu Chao1b,2b, Shang Xiaohong1a,2a, Qiao Yu1a,2a, Li Guimei1a,2a
), Xu Chao1b,2b, Shang Xiaohong1a,2a, Qiao Yu1a,2a, Li Guimei1a,2a
- 1a. Department of Pediatrics; b. Department of Endocrinology and Metabolism, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021,China
2a. Department of Pediatrics; 2b.Department of Endocrinology and Metabolism, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan 250021, China
-
Received:2021-03-10Online:2021-05-20Published:2021-06-09 -
Contact:Sun Yan E-mail:sunyan6150@126.com
CLC Number:
Cite this article
Liu Fan, Sun Yan, Xu Chao, Shang Xiaohong, Qiao Yu, Li Guimei. Clinical characteristics of hereditary renal tubular disease in hypokalemia[J]. Clinical Focus, 2021, 36(5): 436-441.
share this article
Add to citation manager EndNote|Ris|BibTeX
URL: https://huicui.hebmu.edu.cn/EN/10.3969/j.issn.1004-583X.2021.05.010
| 疾病种类 | 例数 | 性别(例) | 百分比(%) | |
|---|---|---|---|---|
| 男 | 女 | |||
| Bartter综合征 | 23 | 15 | 8 | 35.40 |
| 肾小管酸中毒 | 14 | 2 | 12 | 21.54 |
| Gitelman综合征 | 8 | 5 | 3 | 12.31 |
| 不明原因 | 5 | 2 | 3 | 7.69 |
| 糖尿病 | 3 | 2 | 1 | 4.62 |
| 肿瘤 | 2 | 1 | 1 | 3.08 |
| 范可尼综合征 | 2 | 1 | 1 | 3.08 |
| 甲基丙二酸血症 | 2 | 0 | 2 | 3.08 |
| 原发性醛固酮综合征 | 1 | 1 | 0 | 1.54 |
| 甲状腺功能亢进 | 1 | 0 | 1 | 1.54 |
| 肾损害 | 1 | 1 | 0 | 1.54 |
| 低磷性佝偻病 | 1 | 0 | 1 | 1.54 |
| Cushing病 | 1 | 0 | 1 | 1.54 |
| 高苷酸尿症 | 1 | 0 | 1 | 1.54 |
| 疾病种类 | 例数 | 性别(例) | 百分比(%) | |
|---|---|---|---|---|
| 男 | 女 | |||
| Bartter综合征 | 23 | 15 | 8 | 35.40 |
| 肾小管酸中毒 | 14 | 2 | 12 | 21.54 |
| Gitelman综合征 | 8 | 5 | 3 | 12.31 |
| 不明原因 | 5 | 2 | 3 | 7.69 |
| 糖尿病 | 3 | 2 | 1 | 4.62 |
| 肿瘤 | 2 | 1 | 1 | 3.08 |
| 范可尼综合征 | 2 | 1 | 1 | 3.08 |
| 甲基丙二酸血症 | 2 | 0 | 2 | 3.08 |
| 原发性醛固酮综合征 | 1 | 1 | 0 | 1.54 |
| 甲状腺功能亢进 | 1 | 0 | 1 | 1.54 |
| 肾损害 | 1 | 1 | 0 | 1.54 |
| 低磷性佝偻病 | 1 | 0 | 1 | 1.54 |
| Cushing病 | 1 | 0 | 1 | 1.54 |
| 高苷酸尿症 | 1 | 0 | 1 | 1.54 |
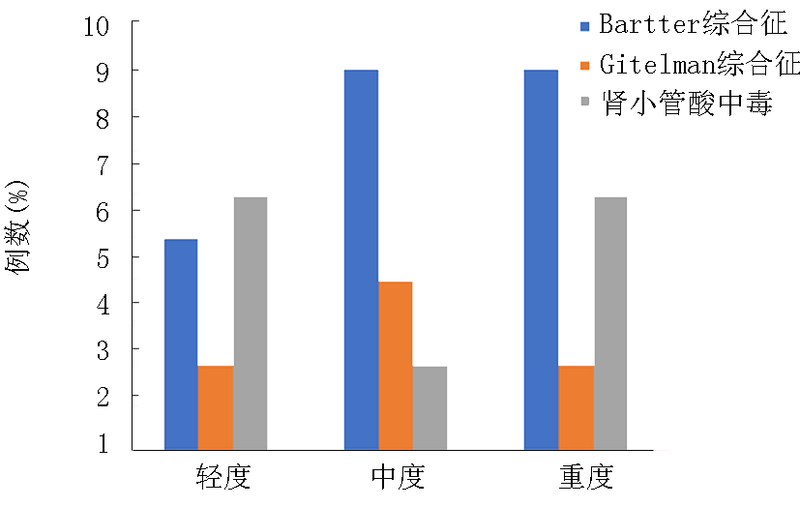
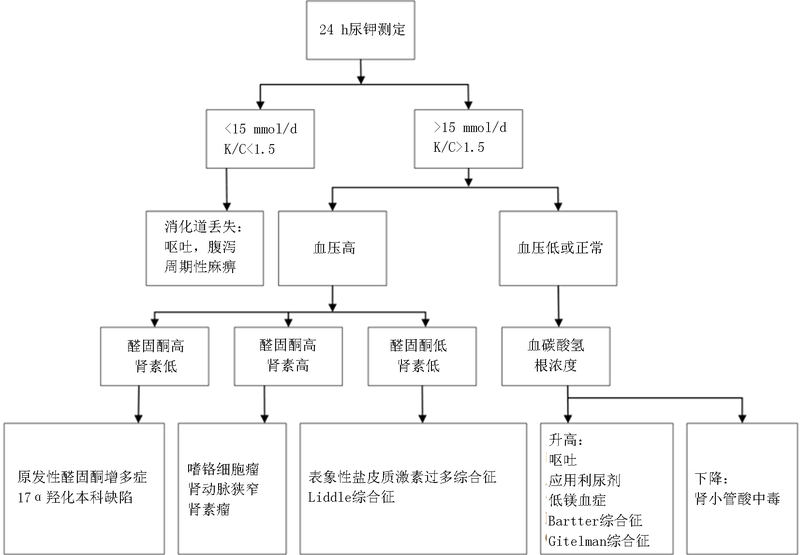
| 项目 | Bartter综合征(n=23) | Gitelman综合征(n=8) | 肾小管酸中毒(n=14) | F值 | P值 |
|---|---|---|---|---|---|
| 年龄(岁) | 3.21±3.69 | 9.75±3.62 | 4.56±4.40 | - | - |
| 性别/(男/女,例) | 15/8 | 5/3 | 2/12 | 5.813 | 0.0059 |
| K+(mmol/L) | 2.58±0.65 | 2.74±0.34 | 2.73±0.56 | 0.7007 | 0.5019 |
| Ca2+(mmol/L) | 2.58±0.18 | 2.33±0.52 | 2.24±0.47 | 0.02061 | 0.9796 |
| Mg2+(mmol/L) | 0.94±0.26 | 0.66±0.08 | 0.99±0.34 | 2.37 | 0.1231 |
| Cl+(mmol/L) | 84.3±14.01 | 96.88±3.04 | 113.57±6.02 | 18.6 | 0.0001 |
| Na+(mmol/L) | 130.97±7.32 | 138.75±2.19 | 136.06±8.38 | 0.5989 | 0.5581 |
| Cr(mmol/L) | 30.92±16.23 | 31.67±10.08 | 36.37±10.57 | 0.1138 | 0.8930 |
| BUN(mmol/L) | 4.06±2.27 | 4.44±1.57 | 3.66±1.66 | 1.230 | 0.3117 |
| 醛固酮(pg/ml) | 425.39±391.99 | 153.50±26.23 | - | - | - |
| 肾素(pg/ml) | 323.49±460.10 | 283.8±226.02 | - | - | - |
| 收缩压(mmHg) | 92.25±11.47 | 106.71±15.70 | 96.22±12.50 | 2.128 | 0.1618 |
| 舒张压(mmHg) | 62.67±7.58 | 67.14±8.53 | 61.89±3.55 | 2.168 | 0.1571 |
| CO2(mmol/L) | 28.69±6.65 | 29.81±4.66 | 12.88±2.37 | 10.45 | 0.0002 |
| 项目 | Bartter综合征(n=23) | Gitelman综合征(n=8) | 肾小管酸中毒(n=14) | F值 | P值 |
|---|---|---|---|---|---|
| 年龄(岁) | 3.21±3.69 | 9.75±3.62 | 4.56±4.40 | - | - |
| 性别/(男/女,例) | 15/8 | 5/3 | 2/12 | 5.813 | 0.0059 |
| K+(mmol/L) | 2.58±0.65 | 2.74±0.34 | 2.73±0.56 | 0.7007 | 0.5019 |
| Ca2+(mmol/L) | 2.58±0.18 | 2.33±0.52 | 2.24±0.47 | 0.02061 | 0.9796 |
| Mg2+(mmol/L) | 0.94±0.26 | 0.66±0.08 | 0.99±0.34 | 2.37 | 0.1231 |
| Cl+(mmol/L) | 84.3±14.01 | 96.88±3.04 | 113.57±6.02 | 18.6 | 0.0001 |
| Na+(mmol/L) | 130.97±7.32 | 138.75±2.19 | 136.06±8.38 | 0.5989 | 0.5581 |
| Cr(mmol/L) | 30.92±16.23 | 31.67±10.08 | 36.37±10.57 | 0.1138 | 0.8930 |
| BUN(mmol/L) | 4.06±2.27 | 4.44±1.57 | 3.66±1.66 | 1.230 | 0.3117 |
| 醛固酮(pg/ml) | 425.39±391.99 | 153.50±26.23 | - | - | - |
| 肾素(pg/ml) | 323.49±460.10 | 283.8±226.02 | - | - | - |
| 收缩压(mmHg) | 92.25±11.47 | 106.71±15.70 | 96.22±12.50 | 2.128 | 0.1618 |
| 舒张压(mmHg) | 62.67±7.58 | 67.14±8.53 | 61.89±3.55 | 2.168 | 0.1571 |
| CO2(mmol/L) | 28.69±6.65 | 29.81±4.66 | 12.88±2.37 | 10.45 | 0.0002 |
| 组别 | 例数 | 胃肠道症状 | 生长迟缓 | 多饮多尿 | 四肢乏力 | 消瘦 | 发热 |
|---|---|---|---|---|---|---|---|
| Bartter综合征 | 23 | 2(8.7) | 6(26.1) | 8(34.8) | 2(8.7) | 1(4.3) | 4(17.4) |
| Gitelman综合征 | 8 | 0 | 4(50.0) | 1(12.5) | 3(37.5) | 0 | 0 |
| 肾小管酸中毒 | 14 | 2(14.3) | 9(64.3) | 1(7.1) | 2(14.3) | 0 | 0 |
| 组别 | 例数 | 胃肠道症状 | 生长迟缓 | 多饮多尿 | 四肢乏力 | 消瘦 | 发热 |
|---|---|---|---|---|---|---|---|
| Bartter综合征 | 23 | 2(8.7) | 6(26.1) | 8(34.8) | 2(8.7) | 1(4.3) | 4(17.4) |
| Gitelman综合征 | 8 | 0 | 4(50.0) | 1(12.5) | 3(37.5) | 0 | 0 |
| 肾小管酸中毒 | 14 | 2(14.3) | 9(64.3) | 1(7.1) | 2(14.3) | 0 | 0 |
| 疾病 | 遗传 方式 | 病例 | OMIM | 突变基因 | 突变位点 蛋白 | 核苷酸改变 | 氨基酸改变 | 致病性 |
|---|---|---|---|---|---|---|---|---|
| Bartter综合征 | AR | A | 613090 | CLCNKB | C1C-Kb | 1-19号外显子纯合缺失 | - | P |
| AR | B | 613090 | CLCNKB | C1C-Kb | CLCNKB基因纯合突变 | - | - | |
| AR | C | 613090 | CLCNKB | C1C-Kb | CLCNKB基因纯合大片段缺失 | - | P | |
| AR | D | 613090 | 阴性 | - | - | - | - | |
| Gitelman综合征 | AR | E | 263800 | SLC12A3 | NCCT | c.1077c>G; c.1456G>A | p.N359K; p.D486N | P;P |
| AR | F | 263800 | SLC12A3 | NCCT | c.578_580del;c.1567G>A | p.193_194del; p.A523T | LP;P | |
| AR | G | 263800 | SLC12A3 | NCCT | c.473G>A; c.1456G>A | p.R158Q; p.D486N | P;P | |
| AR | H | 263800 | SLC12A3 | NCCT | c.783delinsTCATTGGCGTGGTCTCG;c.1698C>A; c.2542G>A | p.1262Sfs*46; p.N566K; p.D848N | LP;VUS; LP | |
| AR | I | 263800 | SLC12A3 | NCCT | c.1367delT | p.L456fs | P | |
| 高苷氨酸尿症 | AR | J | 242600 | SLC6A20 | SIT1 | c.199C>T | p.Q67X | LP |
| 疾病 | 遗传 方式 | 病例 | OMIM | 突变基因 | 突变位点 蛋白 | 核苷酸改变 | 氨基酸改变 | 致病性 |
|---|---|---|---|---|---|---|---|---|
| Bartter综合征 | AR | A | 613090 | CLCNKB | C1C-Kb | 1-19号外显子纯合缺失 | - | P |
| AR | B | 613090 | CLCNKB | C1C-Kb | CLCNKB基因纯合突变 | - | - | |
| AR | C | 613090 | CLCNKB | C1C-Kb | CLCNKB基因纯合大片段缺失 | - | P | |
| AR | D | 613090 | 阴性 | - | - | - | - | |
| Gitelman综合征 | AR | E | 263800 | SLC12A3 | NCCT | c.1077c>G; c.1456G>A | p.N359K; p.D486N | P;P |
| AR | F | 263800 | SLC12A3 | NCCT | c.578_580del;c.1567G>A | p.193_194del; p.A523T | LP;P | |
| AR | G | 263800 | SLC12A3 | NCCT | c.473G>A; c.1456G>A | p.R158Q; p.D486N | P;P | |
| AR | H | 263800 | SLC12A3 | NCCT | c.783delinsTCATTGGCGTGGTCTCG;c.1698C>A; c.2542G>A | p.1262Sfs*46; p.N566K; p.D848N | LP;VUS; LP | |
| AR | I | 263800 | SLC12A3 | NCCT | c.1367delT | p.L456fs | P | |
| 高苷氨酸尿症 | AR | J | 242600 | SLC6A20 | SIT1 | c.199C>T | p.Q67X | LP |
| [1] |
Xu T, Zhu W, Wang P. Cervical ganglioneuroma: A case report and review of the literature[J]. Medicine (Baltimore), 2019, 98(15):e15203.
doi: 10.1097/MD.0000000000015203 URL |
| [2] | Kardalas E, Paschou SA, Anagnostis P, et al. Hypokalemia: A clinical update[J]. Endocr Connect, 2018, 7(4):135-146. |
| [3] |
Pelletier J, Gbadegesin R, Staples B. Renal tubular acidosis[J]. Pediatr Rev, 2017, 38(11):537-539.
doi: 10.1542/pir.2016-0231 pmid: 29093127 |
| [4] |
Lim AK, Choi MJ. Distal renal tubular acidosis associated with Sjogren syndrome[J]. Intern Med J, 2013, 43(12):1330-1334.
doi: 10.1111/imj.12300 pmid: 24330363 |
| [5] | Shahbaz A, Shahid MF, Saleem HMK, et al. Hypokalemic paralysis secondary to renal tubular acidosis revealing underlying Sjogren's Syndrome[J]. Cureus, 2018, 10(8):e3128. |
| [6] |
Amirlak I, Dawson KP. Bartter syndrome: An overview[J]. QJM, 2000, 93(4):207-215.
doi: 10.1093/qjmed/93.4.207 URL |
| [7] |
Cunha TDS, Heilberg IP. Bartter syndrome: Causes, diagnosis, and treatment[J]. Int J Nephrol Renovasc Dis, 2018, 11:291-301.
doi: 10.2147/IJNRD URL |
| [8] | 赵金丽. 肾脏电压门控氯离子通道研究进展[J]. 国际儿科学杂志, 2012, 39(5):477-479. |
| [9] | Besouw MTP, Kleta R, Bockenhauer D. Bartter and Gitelman syndromes: Questions of class[J]. Pediatr Nephrol, 2019, 29:10. |
| [10] |
Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome:consensus and guidance from a Kidney Disease:Improving Global Outcomes (KDIGO) Controversies Conference[J]. Kidney Int, 2017, 91:24-33.
doi: S0085-2538(16)30602-0 pmid: 28003083 |
| [11] |
Walsh PR, Tse Y, Ashton E, et al. Clinical and diagnostic features of Bartter and Gitelman syndromes[J]. Clin Kidney J, 2018, 11(3):302-309.
doi: 10.1093/ckj/sfx118 URL |
| [12] |
Wongsaengsak S, Vidmar AP, Addala A, et al. A novel SLC12A1 gene mutation associated with hyperparathyroidism, hypercalcemia, nephrogenic diabetes insipidus, and nephrocalcinosis in four patients[J]. Bone, 2017, 97:121-125.
doi: S8756-3282(17)30011-X pmid: 28095294 |
| [13] |
Mayr B, Glaudo M, Schöfl C. Activating calcium-sensing receptor mutations: Prospects for future treatment with calcilytics[J]. Trends Endocrinol Metab, 2016, 27(9):643-652.
doi: 10.1016/j.tem.2016.05.005 URL |
| [14] |
Viering DH, de Baaij JH, Walsh SB, et al. Genetic causes of hypomagnesemia,a clinical over[J]. Pediatr Nephrol, 2017, 32(7):1123-1135.
doi: 10.1007/s00467-016-3416-3 pmid: 27234911 |
| [15] |
Wang F, Shi C, Cui Y, et al. Mutation profile and treatment of Gitelman syndrome in Chinese patients[J]. Clin Exp Nephrol, 2017, 21(2):293-299.
doi: 10.1007/s10157-016-1284-6 pmid: 27216017 |
| [16] |
Seyberth HW, Weber S, Komhoff M. Bartter’s and Gitelman’s syndrome[J]. Curr Opin Pediatr, 2017, 29(2):179-186.
doi: 10.1097/MOP.0000000000000447 URL |
| [17] |
Mumford E, Unwin RJ, Walsh SB. Liquorice, Liddle, Bartter or Gitelman-how to differentiate?[J]. Nephrol Dial Transplant, 2019, 34(1):38-39.
doi: 10.1093/ndt/gfy199 URL |
| [18] |
Fulchiero R, Seo-Mayer P. Bartter syndrome and Gitelman syndrome[J]. Pediatr Clin North Am, 2019, 66(1):121-134.
doi: 10.1016/j.pcl.2018.08.010 URL |
| [19] |
Bao M, Cai J, Yang X, et al. Genetic screening for Bartter syndrome and Gitelman syndrome pathogenic genes among individuals with hypertension and hypokalemia[J]. Clin Exp Hypertens, 2019, 41(4):381-388.
doi: 10.1080/10641963.2018.1489547 URL |
| [20] |
Park E, Cho MH, Hyun HS, et al. Genotype-phenotype analysis in pediatric patients with distal renal tubular acidosis[J]. Kidney Blood Press Res, 2018, 43(2):513-521.
doi: 10.1159/000488698 URL |
| [21] |
Alexander RT, Bitzan M. Renal tubular acidosis[J]. Pediatr Clin North Am, 2019, 66(1):135-157.
doi: 10.1016/j.pcl.2018.08.011 URL |
| [1] | Dong Hui, Huang Wenhui, Zhao Hui, Qian Rui. Adult Bartter syndrome complicated with acute exacerbation of chronic renal insufficiency: A case report [J]. Clinical Focus, 2023, 38(11): 1022-1026. |
| [2] | Pang Shu, Zhang Mingkai, Bai Hongmei, Wu Yongdong. Congenital insensitivity to pain with anhidrosis: A case report and literature review [J]. Clinical Focus, 2023, 38(1): 64-67. |
| [3] | Tong Tian, Wang Ruiying, Zhang Lihui, Liu Zhihong, Luo Jianqin, Zhao Zhansheng. Gitelman syndrome with normomagnesemia: A case report and literature review [J]. Clinical Focus, 2022, 37(11): 1031-1036. |
| [4] | Eamran Hossain, Tian Ya, Chen Yuan, Zhang Shaodan, Zhang Huifeng. IPEX syndrome concurrent with gut-origin sepsis: A child patient report and literature review [J]. Clinical Focus, 2021, 36(5): 453-457. |
| [5] | Xu Yuping, Yu Yu, Wang Tuanjie, Li Shujun. A case report and literature review:ornithine transcarbamylase deficiency caused by mutation of c.1006-3c>G#br# [J]. Clinical Focus, 2019, 34(9): 850-854. |
| [6] | Xu Jinteng1, Ma Kai2, Li Xiaoqing2, Yang Ying3. Clinical analysis of Gitelman syndrome and short stature with SLC12A3 mutation in one case [J]. Clinical Focus, 2019, 34(10): 933-936. |
| [7] | Zhang Wei1,2, Zhang Mei1, Wang Yucheng1, Cui Dai1, Zhou Hongwen1,. Pseudohypoparathyroidism and hypokalemia: clinical manifestation analysis [J]. Clinical Focus, 2017, 32(9): 759-762. |
| [8] | Wang Minghui, Bu Haiwei, Li Chunhua, Zhang Ying, Zhao Jie,Sun Wanglexian. Impact of hypokalemia on longterm prognosis in patients with acute myocardial infarction [J]. Clinical Focus, 2016, 31(9): 978-982. |
| [9] | Li Yuej un;Tian Xiang;Geng Wei;Wang Peij un;Zhang Tongle;Zhang Qi;Fu Yongqi. Association of serum potassium level with infarct area and coronary Gensini score in patients with acute myocardial infarction [J]. Clinical Focus, 2015, 30(3): 276-279. |
| [10] | ZHANG Lei;ZHOU Dan;XU Xiao-heng;ZHAO Hui;ZHANG Si-jin. Survey on carrier rates of six common SLC25A13 gene mutations in children of Jilin Province [J]. Clinical Focus, 2014, 29(7): 737-739. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||