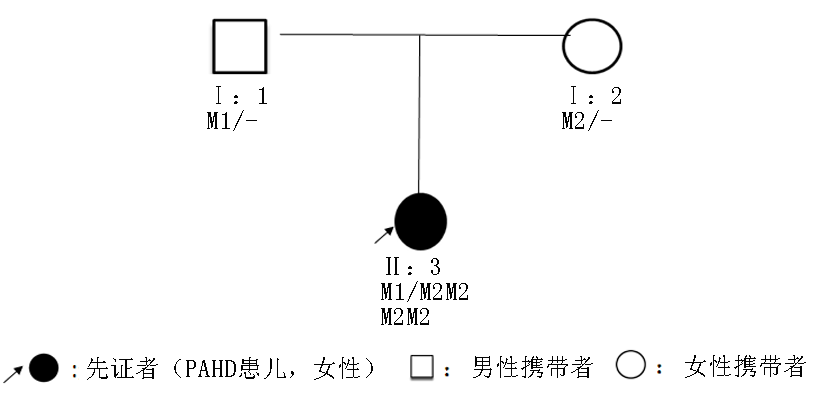
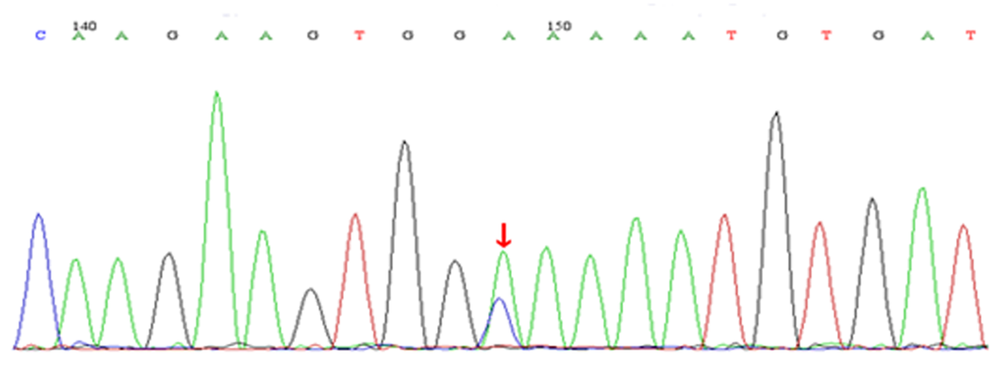
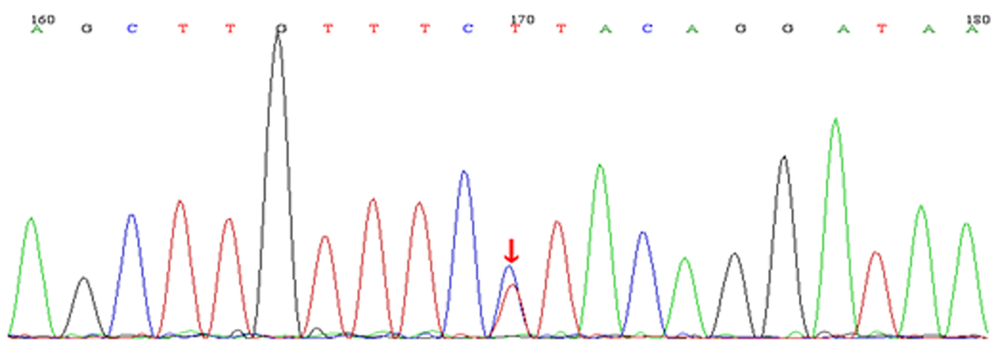
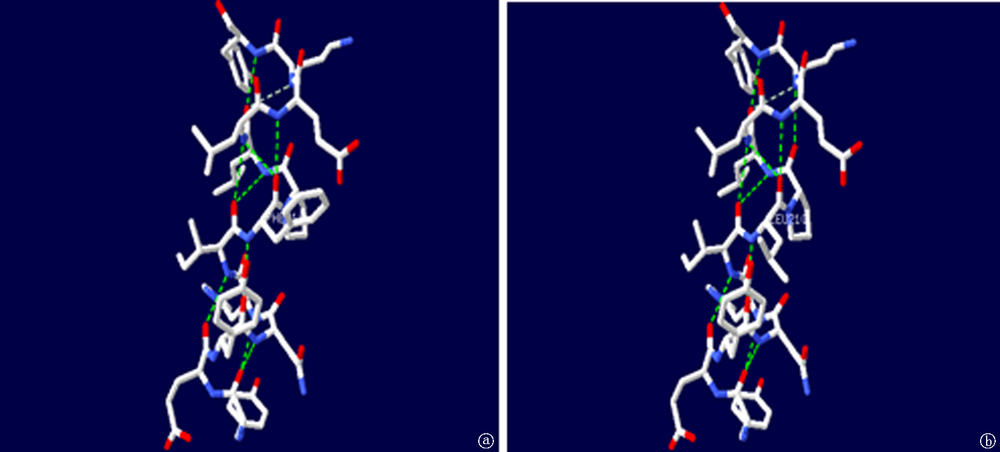
| [1] |
顾学范. 临床遗传代谢病[M]. 北京: 人民卫生出版社, 2015.
|
| [2] |
Ye J, Yang Y, Yu W, et al. Demographics, diagnosis and treatment of 256 patients with tetrahydrobiopterin deficiency in mainland China: Results of a retrospective, multicentre study[J]. J Inherit Metab Dis, 2013, 36(5):893-901.
doi: 10.1007/s10545-012-9550-6
URL
|
| [3] |
叶军, 顾学范. “高苯丙氨酸血症的诊治共识”解读[J]. 中华儿科杂志, 2014, 52(6):430-432.
|
| [4] |
Chen T, Xu W, Wu D, et al. Mutational and phenotypic spectrum of phenylalanine hydroxylase deficiency in Zhejiang Province, China[J]. Sci Rep, 2018, 8(1): 17137.
doi: 10.1038/s41598-018-35373-9
URL
|
| [5] |
Yan Y, Zhang C, Jin X, et al. Mutation spectrum of PAH gene in phenylketonuria patients in Northwest China: Identification of twenty novel variants[J]. Metab Brain Dis, 2019, 34(3):733-745.
doi: 10.1007/s11011-019-0387-7
URL
|
| [6] |
Zhu T, Qin S, Ye J, et al. Mutational spectrum of phenylketonuria in the Chinese Han Population: A novel insight into the geographic distribution of the common mutations[J]. Pediatr Res, 2010, 67:280-285.
doi: 10.1203/PDR.0b013e3181c9fb85
URL
|
| [7] |
Song L, Dang L, Meng Y, et al. Mutation spectrum of phenylalanine hydroxylase gene in patients with phenylketonuria in Tianjin and surrounding areas of Northern China[J]. Chin J Med Genet, 2010, 27(1):7-12.
|
| [8] |
Yan Y, Jin X, Wang X, et al. Screening of PAH common mutations in Chinese phenylketonuria patients using iPLEX MALDI-TOF MS[J]. ACS OMEGA, 2020, 5:1805-1812.
doi: 10.1021/acsomega.9b02955
URL
|
| [9] |
王杰, 朱博, 张丽春, 等. 苯丙氨酸羟化酶基因c.158G>A(p.Arg53His)突变患儿随访及突变位点功能评估[J]. 浙江大学学报:医学版, 2021, 50(4):444-453.
|
| [10] |
张璋, 张立琴, 杜玮, 等. 苯丙氨酸羟化酶缺乏症患儿基因型和表型关系及其临床应用的研究[J]. 临床儿科杂志, 2020, 38(9):671-678.
|
| [11] |
Luleyap U, Pazarci P, Comertpay G, et al. The analysis of the phenylalanine hydroxylase gene mutations by sequencing and ARMS techniques in Turkish patients[J]. Cukurova Med J, 2016, 41(4): 702-708.
doi: 10.17826/cutf.254199
URL
|
| [12] |
Liu N, Huang Q, Li Q, et al. Spectrum of PAH gene variants among a population of Han Chinese patients with phenylketonuria from Northern China[J]. BMC Med Genet, 2017, 18(1): 1-7.
doi: 10.1186/s12881-016-0364-5
URL
|
| [13] |
杜玮, 杨桂芸, 陆薇冰, 等. 青岛市苯丙氨酸羟化酶缺乏症患儿基因突变分析[J]. 发育医学电子杂志, 2020, 8(1):24-28.
|
| [14] |
李菲, 张雪纯, 张德山, 等. PKU患者PAH基因突变分析[J]. 中西医结合心血管病杂志. 2019, 7(2):186-188.
|
| [15] |
鲁程绯, 郭志义, 鲁弼嘉, 等. 唐山市苯丙酮尿症患儿筛查及PAH基因突变情况分析[J]. 天津医药, 2020, 48(10):1006-1009.
|
| [16] |
胡海利, 李卫东, 邵子瑜, 等. 72例安徽地区高苯丙氨酸血症患儿苯丙氨酸羟化酶基因变异观察[J]. 山东医药, 2018, 58(3):70-72.
|
| [17] |
张亚果, 叶飘, 欧明才, 等. 四川省部分地区43例高苯丙氨酸血症患儿相关基因突变分析[J]. 中国儿童保健杂志, 2020, 28(11):1255-1258, 1262.
|
| [18] |
王秋菊, 沈亦平, 陈少科, 等. 遗传变异分类标准与指南[J]. 中国科学:生命科学, 2017, 47(6):668-688.
|
| [19] |
张展, 刘昕, 高峻峻, 等. 83例苯丙酮尿症患者pah基因大片段缺失的检测[J]. 重庆医学, 2018, 47(34):4374-4378.
|
| [20] |
杨艳玲, 叶军. 高苯丙氨酸血症的诊治共识[J]. 中华儿科杂志, 2014, 52(6):420-425.
|
| [21] |
Lee SG, Yim YS, Lee YH, et al. Fasting serum amino acids concentration is associated with insulin resistance and proinflammatory cytokines[J]. DIABETES RES CLIN PR, 2018, 140:107-117.
doi: 10.1016/j.diabres.2018.03.028
URL
|
 )
)